IL-4在支气管哮喘中作用的相关研究
2018/01/15
穆庆
河南省人民医院呼吸与危重症医学科 450003
变应性哮喘是以Th2型细胞为主导的气道慢性炎症性疾病,也是一种遗传性免疫反应紊乱。哮喘的特征是气道高反应性和和肺部炎症,气道高反应性导致气道的收缩,肺部炎症以嗜酸性粒细胞和Th2型淋巴细胞浸润为表现。已经公认,变应性疾病和哮喘包含复杂的程度不等基因紊乱,许多基因研究把哮喘的易感性基因定位在5号染色体长臂2区3带3亚带3次亚带(5q23-33),这个区域包含Th2型细胞因子基因,如IL-4、-5、-9和13[1]。它们的来源多样,可以由肥大细胞、嗜碱性粒细胞和嗜酸性粒细胞产生,也可以由抗原刺激的CD4+T细胞亚群,如Th2和Th9淋巴细胞产生,还可以由细胞因子刺激的原始淋巴母细胞如neucytes产生[2]。IL-4和其他Th型细胞因子一起,在哮喘中发挥着重要的作用:为B细胞IgE表型转换所必需(增加IgE的产生);促进T细胞增殖、决定Th细胞的分化方向;募集嗜酸性粒细胞;促进肥大细胞、杯状细胞增殖(Figure 1)。IL-4作为最重要的Th2型细胞因子,通过IL-4受体α(IL-4 Receptor α,IL-4Rα)及下游的信号传导通路来发挥其生物学活性。哮喘作为一种发病率较高的一种疾病,其治疗也是人们关注的焦点,随着哮喘发病机制的深入研究,越来越多的人把目光放在了以Th2细胞因子为靶向的药物上来。本篇文章就IL-4在哮喘发病、信号转导及分子靶向治疗几个方面作一阐述。河南省人民医院呼吸与危重症医学科 450003
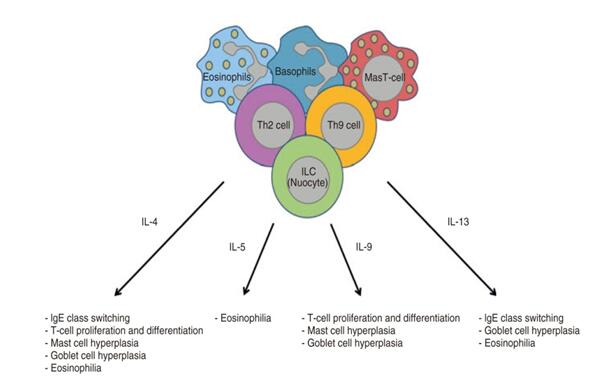
图 1 Th2型效应细胞因子的表达和功能。
Figure 1 Expression and function of type 2 effector cytokines。摘自reference[2]。
Figure 1 Expression and function of type 2 effector cytokines。摘自reference[2]。
1、IL-4对哮喘参与细胞的影响
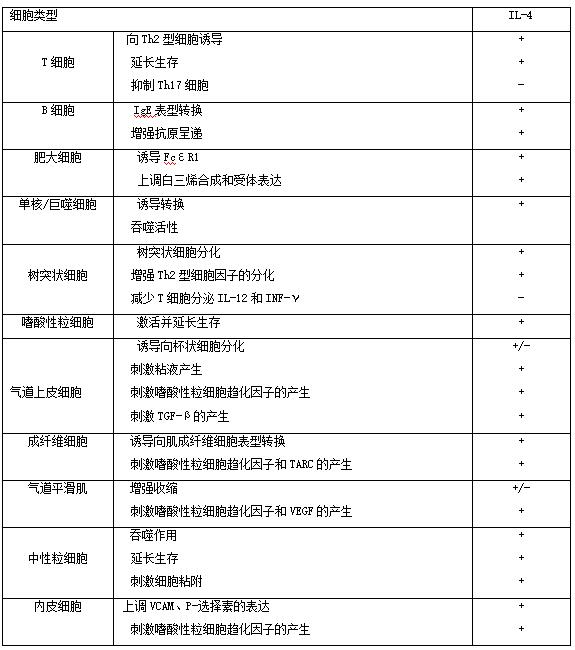
变应原由树突状细胞核巨噬细胞等抗原呈递细胞呈递给CD4+Th2型细胞,Th2型细胞被激活并释放各种白介素,如IL-4、-5、-9和-13。IL-4作用于B细胞刺激其产生IgE,IgE结合于肥大细胞表面。当随后变应原与肥大细胞表面的IgE结合时,肥大细胞就会被激活,并释放IL-4、IL-5、组胺、白三烯和前列腺素等效应物质,募集包括嗜酸性粒细胞、嗜碱性粒细胞、肥大细胞和杯状细胞的增殖,引起支气管平滑肌收缩、黏液分泌、气道嗜酸性粒细胞浸润、气道粘膜充血水肿等,患者即表现为哮喘发作。IL-4作为典型Th2型细胞因子,在哮喘的发病机制中扮演重要的角色,影响着各种各样的细胞(表格1):
1.1IL-4调节B细胞的生长、IgE的产生及膜抗原的表达
1982年,IL-4作为T细胞源性生长因子能够促进B细胞生长第一次被报道[3]。IL-4做为B细胞的转分化因子,通过与B细胞表面的CD40的交叉联系能够促进静止B细胞的DNA合成,诱导B细胞向IgG1和IgE表型的转换。体外实验表明B细胞的分裂数目依赖其膜抗原CD40的配体CD40L的剂量,受IL-4刺激剂量的调节[4]。IL-4能够促进抗IgM预先刺激的B细胞的增殖,这一效应已被作为检测IL-4的生物学活性。IL-4促进B细胞MHCⅡ类抗原和CD40的表达,并增强B细胞提呈抗原能力,使免疫系统能够对小量抗原刺激发生免疫应答,增加FcεRⅡ/CD23(IgE Fc段低亲和力受体)的表达;此外,IL-4还可促进休止期B细胞的早期活化,从G0期进入G1期,细胞体积增大,并表达CD25[5]。
1.2 IL-4对T细胞的作用
IL-4作为典型的Th2型细胞因子,体外实验已经证明在Th2型细胞分化中具有重要的作用[6]。然而,体内研究却发现在IL-4信号缺失的情况下Th2型细胞分化仍能进行[7]。现在认为IL-4信号途径导致GATA3蛋白的上调,在Th2型细胞表达IL-5和-13中具有不可缺少的作用[8]。最近的一项研究也证明了Th2型细胞表达IL-4需要GATA3结合一种顺式作用调节原件(被叫做HS2),位于IL-4基因位点的第二个内含子区,HS2缺陷的T细胞不能表达IL-4,但是IL-5和-13的表达却不受影响[9]。除了IL-4/GATA3-依赖的机制以外,非淋巴来源的TSLP、IL-25(IL-17E)和IL-33对Th2细胞分化也具有重要的作用。Th1型细胞能够分泌IL-2和INF-γ而不能分泌IL-4,反过来,IL-4能够抑制Th1型细胞的增殖并抑制INF-γ的产生。另外,IL-4能够促进细胞毒性T细胞的前体细胞增殖并促进其向细胞毒性T细胞分化[10]。
1.3 IL-4促进杯状细胞增生
哮喘患者过多的粘液分泌是由于杯状细胞数目的增多和粘液腺的肥大[11]。由于气道粘液的堵塞可能危及哮喘患者的生命,所以阐明粘液过度分泌的机制非常重要。关于杯状细胞的起源有两种观点:一种认为起源于表面上皮的非纤毛分泌细胞(Clara细胞),IL-13诱导其发生结构重组并充满粘液[12];另一种观点认为内皮生长因子受体信号和IL-13诱导的STAT6信号通路引起纤毛上皮细胞转分化为非纤毛杯状细胞[13]。一般认为,IL-13和IL-4对杯状细胞的增殖有着非常重要的作用。体外实验表明IL-4或Il-13诱导气道上皮细胞粘液基因的表达[14]。小鼠在体实验也证明IL-4和/或IL-13对变应原诱导的杯状细胞的发展是必需的[12]。
1.4 IL-4对中性粒细胞的影响
哮喘是以Th2型细胞为主导气道慢性炎症性疾病,哮喘肺组织和外周血嗜酸性粒细胞是增加的。但是,在许多哮喘患者气道分泌物中(包括从轻度哮喘到重度哮喘),中性粒细胞是占优势的细胞[15-19]。因此,哮喘气道中性粒细胞增多的现象引起了越来越多的注意。体外实验证明,IL-4对人早幼粒白血病细胞系HL-60的成熟和激活具有重要的作用,也可以增强成熟中性粒细胞的吞噬反应,且这种吞噬能被人IL-4抗体所拮抗[20]。IL-4可以增强中性粒细胞介导的杀菌作用,增强中性粒细胞的呼吸爆发以及调理素介导的吞噬绵羊红细胞作用[21]。IL-4也可以激活中性粒细胞并诱导其CXCL-8的mRNA的表达和炎症因子的释放,例如TNF-α[22]。我们也曾经报道,在马肺动脉内皮细胞,IL-4可以通过增加CXCL-8、E-选择素和VEGF的表达引起中性粒细胞的浸润[23, 24]。但是, IL-4诱导中性粒细胞的增多的在体试验研究少有报道。本论文开始部分的研究证实了IL-4不能直接促进大鼠气道中性粒细胞的募集,然而,IL-4可能通过上调CINC-1和ICAM-1的表达,参与气道中性粒细胞炎症。
1.5 IL-4对其他细胞的影响
上调血管内皮细胞VCAM-1的表达水平;引起变应原诱导的气道嗜酸性粒细胞增多;诱导体外培养的气道平滑肌细胞、肺纤维母细胞、肺泡上皮细胞以及血管内皮细胞嗜酸性粒细胞趋化因子(eotaxins)-1、-2和-3的表达[25, 26];诱导气道上皮细胞eotaxin-1、-2、-3,RANTES、IL-8、gro-α以及MIP-3α的表达[27, 28]。
表格1 IL-4的生物学活性 Table 1 Biological Activities of IL-4.
2、IL-4与哮喘气道高反应(Airway Hyperreactivity,AHR)和气道重塑
2.1 IL-4与AHR
气道高反应性是支气管哮喘的主要特征和诊断依据,表现为气道对各种刺激出现过强或过早的收缩反应。已有文献报道,变异性哮喘患者吸入重组人IL-4后诱导痰中嗜酸性粒细胞增多,气道对乙酰甲胆碱的反应性增高,而且气道反应性的变化与诱导痰中嗜酸性粒细胞的时间变化一致,IL-4可能通过嗜酸性粒细胞的作用来诱导AHR[29]。嗜酸性粒细胞可以分泌主要基础蛋白、嗜酸性粒细胞阳离子蛋白、嗜酸性粒细胞源性神经毒素和嗜酸性粒细胞过氧化物酶、白三烯、血小板激活因子等物质,这些物质与血管渗透性、支气管收缩和气道上皮细胞的破坏密切相关,特别是主要基础蛋白、白三烯和血小板激活因子与气道平滑肌收缩和AHR密切相关[30]。
2.2 IL-4和哮喘气道重塑
哮喘气道重塑与哮喘严重性相关。气道重塑的结构改变包括上皮分离、上皮下纤维化、增生的气道平滑肌细胞聚集、气道平滑肌细胞与上皮间距离缩短、粘液腺和杯状细胞增生、血管增生以及气道粘膜水肿。一般认为,哮喘气道重塑是一个继发的改变,是一种对Th2型炎症的修复反应和炎症持续存在发展的结果。IL-4通过嗜酸性粒细胞、B细胞、肥大细胞、气道上皮细胞介导哮喘的气道重塑。IL-4、-13和粒-巨噬细胞集落刺激因子(Granulocyte-macrophage Colony-stimulating Factor,GM-CSF)影响嗜酸性粒细胞的发生发展,促进其向肺部迁移,而嗜酸性粒细胞通过分泌促纤维化因子转化生长因子(Transforming Growth Factor,TGF)-β引起气道重塑[31]。IL-4和-13促使B细胞发生免疫球蛋白表型转换,即从IgM转换为IgE,当肥大细胞表面表达的FcRI与IgE结合后,肥大细胞被激活,继而发生脱颗粒,脂类介质、各种细胞因子和趋化因子随之释放,介导下游的气道重塑[31]。气道上皮细胞除了作为气道的屏障之外,还是环境刺激物与宿主之间的界面。有新兴的观点认为哮喘患者气道上皮与环境刺激物、感染、炎症刺激物之间的反应是异常的,导致气道上皮某些基因和因子的表达,这些因子会引起上皮下气道重塑[32]。气道重塑的动物模型和人支气管活检标本证明了能够促进气道纤维化和血管生成的上皮来源的细胞因子,包括TGF-β2和VEGF[33, 34]。2010年的一项研究发现骨膜蛋白在哮喘患者支气管上皮细胞的表达式上调的,这种蛋白在TGF-β2信号通路中有重要的作用,还可以促进气道纤维母细胞分泌Ⅰ型胶原蛋白[35]。在IL-4和-13的联合刺激下,哮喘患儿气道上皮表达TGF-β、骨膜蛋白增加[36]。然而,近期的研究发现,在儿童哮喘和疾病的发展中均能观察到气道重塑[37, 38],同时,以气道中性粒细胞炎症(伴随或不伴有嗜酸性粒细胞的浸润)的特征的难治性哮喘也存在着过度的气道重塑[38]。这些研究提示,炎症机制和非炎症机制可能是哮喘气道重塑两个并行的或相互影响的因素。
3、IL-4R信号转导通路
大量研究已经证实IL-4R信号通路在哮喘发病机制具有重要的功能。阻断IL-13或敲除编码IL-4Rα或STAT6的基因的小鼠不能诱导出实验性变应性哮喘模型[39, 40]。下面就IL-4R的信号转导通路作一阐述(Figure 2):
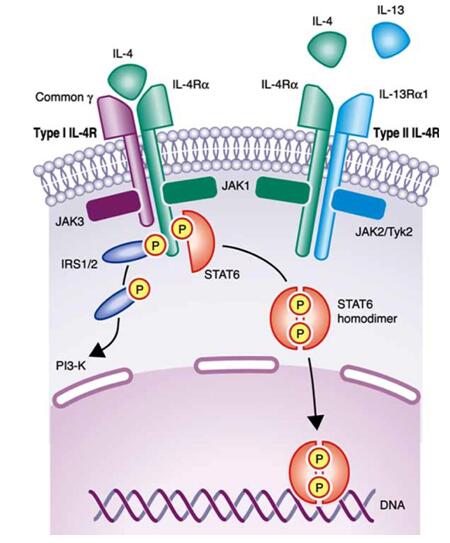
图2 IL-4和-13的信号转导通路
Figure 2 IL-4 and IL-13 signaling pathways.摘自reference[41]。IL-4R有两种类型,Ⅰ型IL-4R和Ⅱ型IL-4R,除了人T细胞、小鼠T细胞和B细胞只表达Ⅰ型IL-4R外,大多数细胞两种受体均表达。Ⅰ型IL-4R由IL-4Rα和普通γ链组成,而Ⅱ型IL-4R由IL-4Rα和IL-13Rα1组成,两者共用IL-4Rα亚基。在配体结合方面,Ⅰ型IL-4R可结合IL-4,而Ⅱ型IL-4R可同时结合IL-4和-13。受体结合后使受体相关性janus激酶(JAK)家族成员磷酸化,JAK1与IL-4Rα结合,JAK3与普通γ链结合,JAK2和TyK2与IL-13Rα1结合。受体异质性二聚体激活JAKs通路进而使IL-4Rα胞质部分尾端的酪氨酸残基磷酸化[41]。
IL-4Rα497位酪氨酸周围区域氨基酸序列与胰岛素和胰岛素样生长因子受体有高度的同源性,这个区域被定义为胰岛素/IL-4受体(I4R)域,IL-4Rα497位酪氨酸磷酸化引起胰岛素受体底物(Insulin Receptor Subtrate,IRS)-1和-2的募集磷酸化,磷酸化的IRS-1和-2为下游酶类和转导蛋白提供了结合位点,最后引起磷酸肌醇激酶3(PI3-K)的募集和激活[42]。已有证据表明I4R介导的IRS-2信号传导对变态性气道炎症具有保护作用[43]。
IL-4Rα第575、603和631位酪氨酸磷酸化对转录因子6的信号诱导剂和激活剂(STAT6)单体靠近IL-4Rα非常重要,随后STAT6磷酸化并与IL-4Rα分离,两个STAT6单体在细胞质结合形成STAT6同质二聚体[44]。STAT6同质二聚体进入细胞核并与IL-4或/和IL-13反应基因的启动子结合。在IL-4Rα介导的各种信号传导通路中,STAT6对Th2细胞分化和IgE抗体转换最为重要[45],因此,STAT6对小鼠实验性变异性哮喘的发生发展是必需的[46]。
4、Anti-IL-4在哮喘治疗中的应用:
目前,治疗哮喘的主要措施有吸入激素、β2受体激动剂、白三烯受体拮抗剂、抗组胺药物以及免疫治疗。虽然这些治疗方法对大多数患者有效,但是仍有一部分患者无效。除了免疫治疗,这些治疗措施只是改善哮喘症状,而不是改变疾病发展的过程。然而,新兴的针对IL-4、-13和-9靶向治疗有望成为治疗哮喘的新希望,对哮喘患者产生短期和长期的效益,下面仅对以IL-4为靶向的药物做以阐述。
4.1 体内改变细胞因子活性的途径
可溶性细胞因子受体可以与游离的细胞因子结合而阻止其与靶细胞结合。Immunex公司曾经推出过可溶性IL-4受体,后因大量试验证明对改善肺功能无益停止研究。针对细胞因子受体的抗体可以直接阻断细胞因子的下游效应。因为IL-4和-13公用一个受体亚基,所以针对IL-4Rα链的单克隆抗体的一个独特优点就在于可以同时阻断IL-4和-13的生物学效应。具有拮抗作用的变异配体(细胞因子突变体)也是一个发展方向,IL-4两个氨基酸残基改变后所形成的的复合物Pitrakinra即是一个例子。Pitrakinra与IL-4Rα链有着很高的亲和力,却没有激活IL-4R的活性,同时干扰IL-4和-13的生物学效应。另外一种阻断细胞因子的方法是运用可吸收反义链寡核苷酸(RASONs)阻断细胞因子基因的转录,例如STAT6或IRS-1/-2的拮抗剂可以干扰IL-4R胞内信号的转导。SOCS是天然存在的STAT6抑制剂,SOCS-1可以抑制所有JAK家族成员的激活[47],所以,我们可以开发一种类似SOCS蛋白质,一方面可以结合STAT6,另一方面又无法引起下游效应。
4.2 目前针对IL-4的靶向制品
IL-4的变异体:Pitrakinra是Aerovance公司推出的分子大小为15-kDa的重组IL-4变异体,作为竞争性IL-4和-13受体拮抗剂,其可以与IL-4Rα链结合,通过阻止IL-4Rα与IL-2Rγ或IL-1Rα的组装而阻断信号转导。Pitrakinra改变的两个氨基酸残基为121位的精氨酸变为门冬氨酸、124位的酪氨酸变为门冬氨酸,这两个氨基酸的变异不会影响Pitrakinra与IL-4Rα的亲和力;在前期临床试验中,Pitrakinra可以抑制哮喘猴抗原特异性IgE的产生、抗原诱导的气道高反应性和肺部嗜酸性粒细胞的侵润[48]。2007年的一项临床Ⅱa期研究显示吸入性抗原激发后,无论是皮下还是雾化吸入Pitrakinra都可以改善哮喘患者的肺功能[49]。但是比较皮下用药和吸入用药发现,吸入应用可以产生前后一致的药理学作用,而皮下用药的效果不稳定,这可能与皮下用药时肺部生物利用度降低有关[48]。针对顽固性哮喘患者的临床Ⅱb期研究已在进行,结果尚未报到(Study ID Number: PPD/2007/AER 001 DP1/2b/ Clinicaltrials.gov Identifier: NCT00801853)。
以IL-4Rα为靶向的抗体:这种抗体可以同时抑制IL-4和-13的信号传导。AMG-317是一种针对IL-4Rα的人单克隆IgG2抗体。Ⅰ期临床试验显示了药物的安全性以及一周一次的皮下用药可以达到稳定的血药浓度(Study ID Number: 20060161/Clinicaltrials.gov Identifier: NCT00436670)。294例Ⅱ期随机双盲安慰剂对照试验显示经过12周治疗,患者首要终点指标哮喘控制问卷得分较基线有所改善,但是没有达到显著性水平;同样,次要终点指标也没有达到显著性水平,但是一些次要终点指标有所改善,例如,血清总IgE水平、哮喘急性发作次数;试验中大剂量AMG-317(300mg,1次/周,皮下注射)可以改善FEV1[50]。
以IL-4Rα为靶向的小分子:应用反义mRNA技术开发的药物AIR-645就是针对IL-4Rα亚单位治疗哮喘的吸入性小分子物质。Ⅰ期临床试验显示正常受试对象和控制较好的哮喘患者对此药具有较好的耐受性和安全性(Study ID Numbers: AIR645-CS1,AIR645-CS2/Clinicaltrials.gov Identifier: NCT0000658749, NTC00941577)。Ⅱ期临床试验尚未进行。
综上所述,IL-4作为Th2型细胞因子,在支气管哮喘的发病机制中有着不可代替的作用,通过影响哮喘炎症细胞和气道结构细胞引起Th2型炎症反应以及中性粒细胞炎症,促进支气管哮喘AHR以及气道重塑。针对IL-4R信号传导通路的靶向药物制品正在开发或已进入临床试验。充分认识IL-4的作用机制将有助于支气管哮喘越来越多新药的开发。
参考文献
1. Steinke, J.W., Current prospective of anti-IL-4, -IL-9, and -IL-13 therapies in allergic disease. Recent Pat Inflamm Allergy Drug Discov, 2010. 4(3): p. 222-30.
2. Oliphant, C.J., J.L. Barlow, and A.N. McKenzie, Insights into the initiation of type 2 immune responses. Immunology, 2011. 134(4): p. 378-85.
3. Howard, M., et al., Identification of a T cell-derived b cell growth factor distinct from interleukin 2. J Exp Med, 1982. 155(3): p. 914-23.
4. Hasbold, J., et al., Cell division number regulates IgG1 and IgE switching of B cells following stimulation by CD40 ligand and IL-4. Eur J Immunol, 1998. 28(3): p. 1040-51.
5. Kikutani, H., et al., Molecular structure of human lymphocyte receptor for immunoglobulin E. Cell, 1986. 47(5): p. 657-65.
6. Swain, S.L., et al., IL-4 directs the development of Th2-like helper effectors. J Immunol, 1990. 145(11): p. 3796-806.
7. Jankovic, D., et al., Single cell analysis reveals that IL-4 receptor/Stat6 signaling is not required for the in vivo or in vitro development of CD4+ lymphocytes with a Th2 cytokine profile. J Immunol, 2000. 164(6): p. 3047-55.
8. Zhu, J., et al., Conditional deletion of Gata3 shows its essential function in T(H)1-T(H)2 responses. Nat Immunol, 2004. 5(11): p. 1157-65.
9. Tanaka, S., et al., The enhancer HS2 critically regulates GATA-3-mediated Il4 transcription in T(H)2 cells. Nat Immunol, 2011. 12(1): p. 77-85.
10. Trenn, G., et al., B cell stimulatory factor 1 (IL-4) enhances the development of cytotoxic T cells from Lyt-2+ resting murine T lymphocytes. J Immunol, 1988. 140(4): p. 1101-6.
11. Woodruff, P.G. and J.V. Fahy, Airway remodeling in asthma. Semin Respir Crit Care Med, 2002. 23(4): p. 361-7.
12. Kuperman, D.A., et al., IL-4 receptor signaling in Clara cells is required for allergen-induced mucus production. J Immunol, 2005. 175(6): p. 3746-52.
13. Tyner, J.W., et al., Blocking airway mucous cell metaplasia by inhibiting EGFR antiapoptosis and IL-13 transdifferentiation signals. J Clin Invest, 2006. 116(2): p. 309-21.
14. Atherton, H.C., G. Jones, and H. Danahay, IL-13-induced changes in the goblet cell density of human bronchial epithelial cell cultures: MAP kinase and phosphatidylinositol 3-kinase regulation. Am J Physiol Lung Cell Mol Physiol, 2003. 285(3): p. L730-9.
15. Sur, S., et al., Sudden-onset fatal asthma. A distinct entity with few eosinophils and relatively more neutrophils in the airway submucosa? Am Rev Respir Dis, 1993. 148(3): p. 713-9.
16. Norzila, M.Z., et al., Interleukin-8 secretion and neutrophil recruitment accompanies induced sputum eosinophil activation in children with acute asthma. Am J Respir Crit Care Med, 2000. 161(3 Pt 1): p. 769-74.
17. Fahy, J.V., et al., Prominent neutrophilic inflammation in sputum from subjects with asthma exacerbation. J Allergy Clin Immunol, 1995. 95(4): p. 843-52.
18. Lamblin, C., et al., Bronchial neutrophilia in patients with noninfectious status asthmaticus. Am J Respir Crit Care Med, 1998. 157(2): p. 394-402.
19. Turner, M.O., et al., Exacerbations of asthma without sputum eosinophilia. Thorax, 1995. 50(10): p. 1057-61.
20. Bober, L.A., et al., IL-4 induces neutrophilic maturation of HL-60 cells and activation of human peripheral blood neutrophils. Clin Exp Immunol, 1995. 99(1): p. 129-36.
21. Boey, H., et al., Interleukin-4 is a neutrophil activator. J Allergy Clin Immunol, 1989. 83(5): p. 978-84.
22. Lavoie-Lamoureux, A., et al., IL-4 activates equine neutrophils and induces a mixed inflammatory cytokine expression profile with enhanced neutrophil chemotactic mediator release ex vivo. Am J Physiol Lung Cell Mol Physiol, 2010. 299(4): p. L472-82.
23. Huang, H., et al., IL-4 stimulates the expression of CXCL-8, E-selectin, VEGF, and inducible nitric oxide synthase mRNA by equine pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol, 2007. 292(5): p. L1147-54.
24. Huang, H., A. Lavoie-Lamoureux, and J.P. Lavoie, Cholinergic stimulation attenuates the IL-4 induced expression of E-selectin and vascular endothelial growth factor by equine pulmonary artery endothelial cells. Vet Immunol Immunopathol, 2009. 132(2-4): p. 116-21.
25. Moore, P.E., et al., IL-13 and IL-4 cause eotaxin release in human airway smooth muscle cells: a role for ERK. Am J Physiol Lung Cell Mol Physiol, 2002. 282(4): p. L847-53.
26. Abonyo, B.O., M.S. Alexander, and A.S. Heiman, Autoregulation of CCL26 synthesis and secretion in A549 cells: a possible mechanism by which alveolar epithelial cells modulate airway inflammation. Am J Physiol Lung Cell Mol Physiol, 2005. 289(3): p. L478-88.
27. Meyer-Hoffert, U., et al., Th2- and to a lesser extent Th1-type cytokines upregulate the production of both CXC (IL-8 and gro-alpha) and CC (RANTES, eotaxin, eotaxin-2, MCP-3 and MCP-4) chemokines in human airway epithelial cells. Int Arch Allergy Immunol, 2003. 131(4): p. 264-71.
28. Reibman, J., et al., Airway epithelial cells release MIP-3alpha/CCL20 in response to cytokines and ambient particulate matter. Am J Respir Cell Mol Biol, 2003. 28(6): p. 648-54.
29. Shi, H.Z., et al., Effect of inhaled interleukin-4 on airway hyperreactivity in asthmatics. Am J Respir Crit Care Med, 1998. 157(6 Pt 1): p. 1818-21.
30. Gleich, G.J., The eosinophil and bronchial asthma: current understanding. J Allergy Clin Immunol, 1990. 85(2): p. 422-36.
31. Sumi, Y. and Q. Hamid, Airway remodeling in asthma. Allergol Int, 2007. 56(4): p. 341-8.
32. Holgate, S.T., et al., The role of the airway epithelium and its interaction with environmental factors in asthma pathogenesis. Proc Am Thorac Soc, 2009. 6(8): p. 655-9.
33. Kelly, M.M., et al., Epithelial expression of profibrotic mediators in a model of allergen-induced airway remodeling. Am J Respir Cell Mol Biol, 2005. 32(2): p. 99-107.
34. Torrego, A., et al., Expression and activation of TGF-beta isoforms in acute allergen-induced remodelling in asthma. Thorax, 2007. 62(4): p. 307-13.
35. Sidhu, S.S., et al., Roles of epithelial cell-derived periostin in TGF-beta activation, collagen production, and collagen gel elasticity in asthma. Proc Natl Acad Sci U S A, 2010. 107(32): p. 14170-5.
36. Lopez-Guisa, J.M., et al., Airway epithelial cells from asthmatic children differentially express proremodeling factors. J Allergy Clin Immunol, 2012. 129(4): p. 990-997 e6.
37. Cokugras, H., et al., Ultrastructural examination of bronchial biopsy specimens from children with moderate asthma. Thorax, 2001. 56(1): p. 25-9.
38. Holgate, S.T. and R. Polosa, The mechanisms, diagnosis, and management of severe asthma in adults. Lancet, 2006. 368(9537): p. 780-93.
39. Grunig, G., et al., Requirement for IL-13 independently of IL-4 in experimental asthma. Science, 1998. 282(5397): p. 2261-3.
40. Akimoto, T., et al., Abrogation of bronchial eosinophilic inflammation and airway hyperreactivity in signal transducers and activators of transcription (STAT)6-deficient mice. J Exp Med, 1998. 187(9): p. 1537-42.
41. Kuperman, D.A. and R.P. Schleimer, Interleukin-4, interleukin-13, signal transducer and activator of transcription factor 6, and allergic asthma. Curr Mol Med, 2008. 8(5): p. 384-92.
42. Wurster, A.L., et al., Stat6 and IRS-2 cooperate in interleukin 4 (IL-4)-induced proliferation and differentiation but are dispensable for IL-4-dependent rescue from apoptosis. Mol Cell Biol, 2002. 22(1): p. 117-26.
43. Blaeser, F., et al., Targeted inactivation of the IL-4 receptor alpha chain I4R motif promotes allergic airway inflammation. J Exp Med, 2003. 198(8): p. 1189-200.
44. Ryan, J.J., et al., Growth and gene expression are predominantly controlled by distinct regions of the human IL-4 receptor. Immunity, 1996. 4(2): p. 123-32.
45. Linehan, L.A., et al., STAT6 is required for IL-4-induced germline Ig gene transcription and switch recombination. J Immunol, 1998. 161(1): p. 302-10.
46. Kuperman, D., et al., Signal transducer and activator of transcription factor 6 (Stat6)-deficient mice are protected from antigen-induced airway hyperresponsiveness and mucus production. J Exp Med, 1998. 187(6): p. 939-48.
47. Losman, J.A., et al., Cutting edge: SOCS-1 is a potent inhibitor of IL-4 signal transduction. J Immunol, 1999. 162(7): p. 3770-4.
48. Tomkinson, A., et al., Inhaled vs subcutaneous effects of a dual IL-4/IL-13 antagonist in a monkey model of asthma. Allergy, 2010. 65(1): p. 69-77.
49. Wenzel, S., et al., Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet, 2007. 370(9596): p. 1422-31.
50. Corren, J., et al., A randomized, controlled, phase 2 study of AMG 317, an IL-4Ralpha antagonist, in patients with asthma. Am J Respir Crit Care Med, 2010. 181(8): p. 788-96.
上一篇:
儿童哮喘发作和发病的心理和生活方式危险因素
下一篇:
阻塞性睡眠呼吸暂停低通气综合征与支气管哮喘









