哮喘气道重塑的发生机制和治疗对策
2015/09/24
第四军医大学西京医院呼吸与危重症医学科 710032
支气管哮喘(以下简称“哮喘”)是以可逆性气流受限及气道高反应性为特征的慢性气道炎症性疾患。慢性气道炎症可导致气道结构的改变,即气道重塑。气道重塑被认为是引起不可逆性气流受限和气道高反应性的重要病理改变。因此防止和减轻气道重塑是哮喘治疗的重要策略,也是治疗的难点。近十年来对哮喘气道重塑的发生机制进行了大量研究,取得了一些令人鼓舞的成绩,给预防和治疗带来了希望。本文对近年的相关研究进展进行综述,希望对提高气道重塑和哮喘规范化防治的认识水平有所帮助。
一、气道重塑病理特点、发生机制
1、气道重塑的病理特点
气道重塑是由于反复的损伤和修复所致的气道壁结构变化,其主要改变包括气道上皮完整性的破坏、气道上皮下纤维化导致网状基底膜增厚、细胞外基质沉积、新生血管形成、血管重构、气道平滑肌(ASM)细胞增生肥大、杯状细胞化生和粘液腺增生等(图1)[1]。其中,上皮下纤维化及基底膜增厚(reticular basement membrane, RBM)是哮喘气道重塑重要特征之一。而这些改变主要由CD4+ T细胞、嗜酸性粒细胞、中性粒细胞及肥大细胞等炎性细胞激发的进行性慢性炎症所致(图2)[2]。

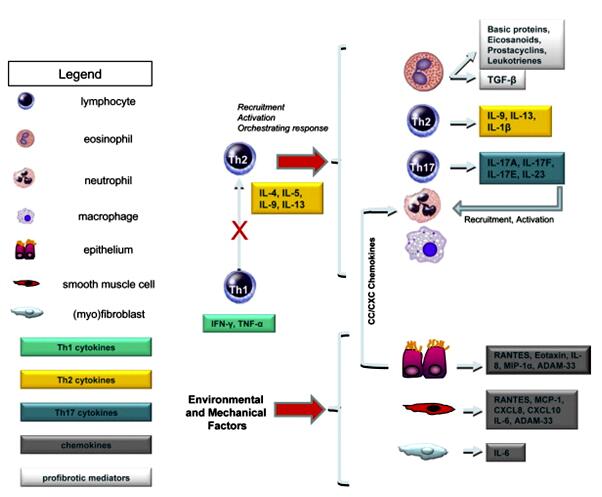
图2.哮喘气道重塑涉及的炎性介质和炎性细胞
气道上皮细胞是机体应对细菌、病毒和环境污染物发生免疫反应和再生反应的重要调节器。气道上皮的损伤在哮喘的发生和炎症的持续发展过程中具有十分重要的地位,是慢性炎症与重塑发生发展的重要病理基础。目前研究认为,气道上皮细胞的病理改变是气道重塑的主要特征[2],在不同程度的哮喘中具有不同的形态学表现[3]。气道上皮细胞的形态改变包括:上皮细胞脱落,纤毛缺失,杯状细胞增生,生长因子、细胞因子、趋化因子表达上调。此外众多研究发现哮喘患者气道上皮的屏障功能是异常的,且存在损伤修复后细胞之间紧密连接的破坏[4,5]。细胞间紧密连接的破坏可使变应原直接作用于抗原提呈细胞,激活免疫反应,诱发过敏性哮喘(图3)[6]。
有证据表明在哮喘中气道上皮更易于受损,且存在异常的修复反应,如EGFR表达增加、表达CDK抑制物p21waf [4,7-8]。当气道上皮损伤后,气道上皮即刻启动修复程序(图3)[9]。气道上皮细胞在对损伤或炎症因子反应修复期间,失去其上皮极性、紧密连接等特点,获得游走和浸袭能力,表达间充质细胞标志,并产生致纤维性介质(如TGF-β、成纤维细胞生长因子、ET-1等)调控成纤维细胞分化和功能,使肌成纤维细胞增多并分泌胶原、弹性蛋白、蛋白多糖等,发生EMT,导致气道壁增厚。目前研究显示EMT是成纤维细胞的一个新的来源在哮喘气道重塑中发挥重要作用。损伤的气道上皮细胞分泌TGF-β,能促进EMT、和杯状细胞增生等(图4)[10,11]。研究显示哮喘患者的气道上皮在对TGF-β的反应中较非哮喘者呈现出更广泛的EMT[12]。研究还显示嗜酸性粒细胞也可通过以下机制参与气道重塑:⑴释放TGF-β; ⑵与肥大细胞相互作用使之合成和释放TGF-β, ⑶激活巨噬细胞合成和释放TGF-β(图5)[13]。
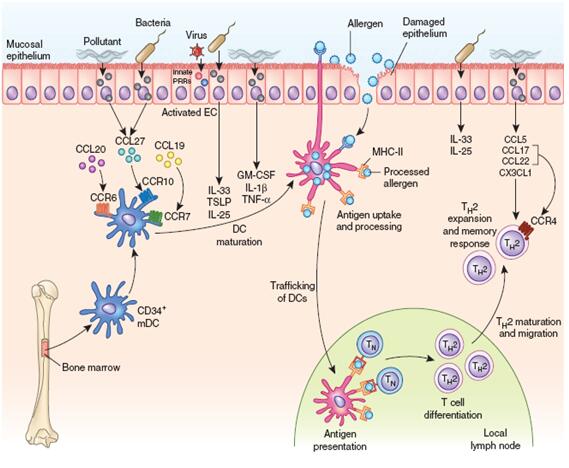
图3. 气道免疫反应的发生过程
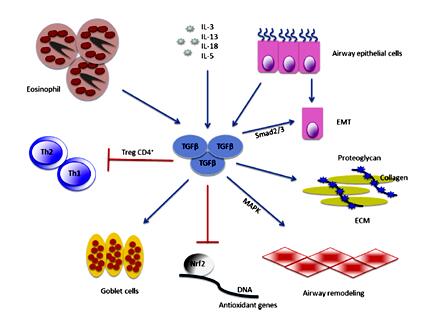
图4. TGF-β在哮喘中的作用

图5.嗜酸性粒细胞诱导EMT体内实验结果
研究表明与非哮喘者相比,致死性哮喘患者ASM增加50%-200%,而非致死性哮喘患者则增加25%-55%,此改变主要由ASM增生或肥大所致(图6)[10]。ASM层增厚是哮喘气道重塑的重要特征,与哮喘的病情严重程度相关,是临床症状和气道高反应性形成的重要基础。哮喘中ASM获得了向气道上皮迁移的能力,是气道重塑的特征之一,它能够突破基底膜的限制,迁移至粘膜下层,导致粘膜下肌化,使得气道壁的增厚程度进一步加剧[14]。此外ASM通过释放前炎性细胞因子、趋化因子、生长因子和ECM蛋白参与气道炎症和重塑的发生和发展。

图6.哮喘气道结构的变化

图7. ASM收缩性的调节
研究显示,哮喘气道假复层柱状纤毛细胞脱落留下基细胞疏松地与基底膜相连,上皮下弹力纤维溶解变性和断裂,纤维短小散在,上皮下网状基底膜明显增厚(图8、9)[18,19]。基底膜增厚主要与气道上皮下过度沉积细胞外基质成分(尤其是胶原纤维)有关。真基底膜(分致密层和疏松层)主要由IV型胶原、粘蛋白和纤维连接蛋白构成,其厚度正常;而基底膜下(网状基底膜)则有III型胶原、Ⅴ型胶原、Ⅰ型胶原、纤维粘连蛋白以及IgG和IgM沉积。业已证实增厚的基底膜主要是基底膜下的增生。而细胞外基质的增加可降低气道顺应性,改变气道平滑肌收缩力方向,使得其收缩所致的气道狭窄更为明显,并与哮喘病情严重度和气道高反应性相关[20]。研究表明上皮下成纤维细和肌纤母细胞是细胞外基质的主要来源。在哮喘患者气道中肌纤维母细胞的数量显著增加,且与网状基底膜的厚度呈正相关。Ⅰ型和III型胶原在上皮下层及平滑肌束内的沉积虽然增加了气道壁厚度、降低了气道壁可扩张性,但同时又能抵抗气腔的进一步缩小,对缓和因哮喘激发因子所致的症状加重和气道反应增高有保护作用[21]。
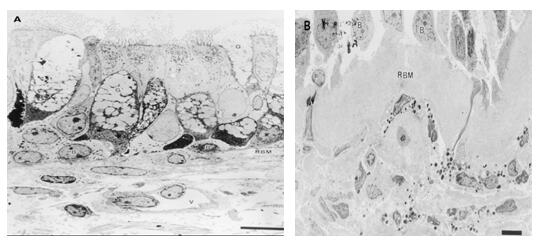
图8.正常对照者和过敏原激发24小时后过敏性哮喘患者粘膜的透射电镜比较

图9.哮喘和正常对照气道弹力纤维系统变化示意图
气道重塑是在气道炎症基础上的异常损伤修复过程,其发生机制复杂几乎涉及气道壁的所有元素,是多种细胞因子、炎性介质、黏附因子等共同作用的结果,参与其调控的信号转导通路极其复杂(图10)。目前证实参与哮喘气道重塑的信号转导通路有NF-κB通路、MAPK通路、PI3K-Akt通路、Rho/ROCK通路、TGF-β1/Smads通路、Wnt/β-catenin通路、Sonic Hedgehog (Shh) 通路等(图11)[22]。其中β-catenin通路在气道重塑的过程中发挥重要作用。

图10.哮喘气道重塑的发生机制

图11 EMT涉及的信号转导通路

图12 .Wnt依赖的β-catenin信号通路
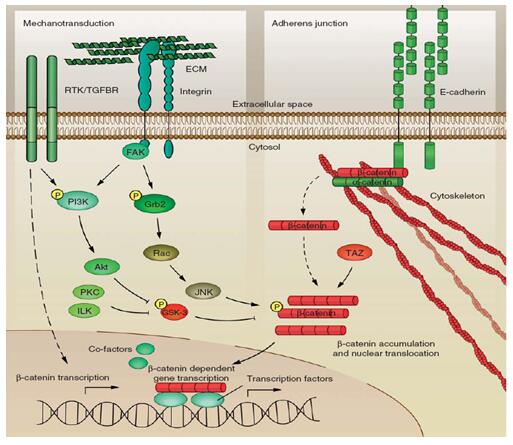
图13. Wnt非依赖的β-catenin信号通路
气道重塑与哮喘临床症状的严重程度、肺功能损害程度存在密切的关联,但目前仍尚无有效的防治措施。虽然许多药物已被证明在动物模型中抑制过敏原诱导的气道重塑,却很少有研究表明能逆转已形成的重塑。近来有研究报道避免抗原接触及糖皮质激素治疗可部分逆转马喘息病(一个自然发生的哮喘样疾病)的ASM重构[24],该结果使我们看到有效治疗气道重塑的一线曙光。
临床试验和动物研究发现,当前用于哮喘治疗的药物如:吸入性糖皮质激素(ICS)、吸入性糖皮质激素联合长效β2受体激动剂、白三烯受体调节剂、抗胆碱能药物、奥马佐单抗等对气道重塑具有一定的抑制作用(表1)。随着研究的不断深入,潜在的治疗靶点不断被发现(表2),极大地丰富了气道重塑的治疗理论,有力的推动了气道重塑治疗药物的研发[25]。目前以β-catenin信号转导通路为靶向研发的药物有:端锚聚合酶抑制剂(Tankyrase inhibitors)、GSK-3抑制剂、核β-catenin和辅因子的相互作用抑制剂[23]。
Tankyrase1、2是一类多聚ADP核糖化酶,有降解axin2的作用,因此使β-catenin 降解复合物失活,从而提各胞浆中的β-catenin水平并,激活β-catenin 信号。其抑制物XAV939体内外证实可抑制博来霉素或TGF- β过表达的腺病毒所致的小鼠皮肤纤维化及博来霉素诱导的小鼠肺纤维化,并可体外抑制TGF- β所诱导的EMT,提示该抑制剂可能对气道重塑也有抑制作用。
GSK-3除降解β-catenin外,对转录因子、激酶和细胞周期调节蛋白等均有调节作用。其抑制剂可抑制哮喘动物模型Eos气道炎和粘液分泌;抑制TGF- β所致的豚鼠肌纤母细胞增殖和IL-1 β诱导的ASMCs 前炎性细胞因子的分泌,说明GSK-3抑制剂可抑制气道重塑。
β-catenin需与Smad2/3或CBP、P300等结合才能发挥转录调节作用。小分子ICG-001(核β-catenin和辅因子的相互作用抑制剂)可阻断β-catenin与CBP的结合,IQ-1(核β-catenin和辅因子的相互作用抑制剂)可阻断β-catenin与P300的结合。以上两种小分子化合物均能阻止核β-catenin发挥转录调节作用,进而抑制气道重塑。
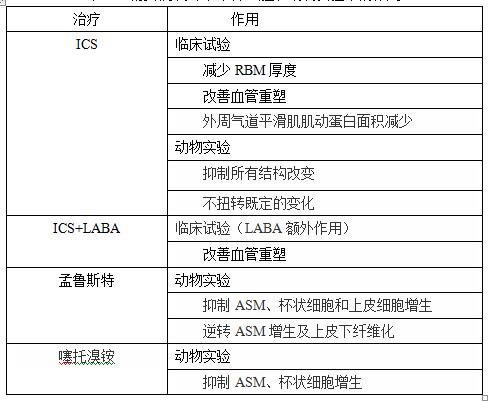
表2 气道重塑的体外研究概括--信号分子作为药物靶点

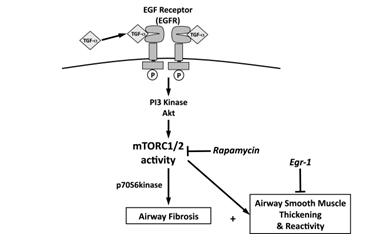
图14 TGF-α Tg/Egr-1ko/ko模型信号通路和表型示意图
综上所述,气道重塑是哮喘的重要病理特征,预防和治疗气道重塑对哮喘防治有重要意义。但目前关于气道重塑的分子机制尚不完全清楚,缺乏对气道重塑有效的治疗药物,相当一部分患者因此发展成不可逆的气流受限,失去了对现有药物治疗的反应性。因此预防和治疗气道重塑对减轻症状,改善生活质量和劳动能力有着十分重要的临床意义。
参考文献
[1] Bergeron C, Tulic MK, Hamid Q. Airway remodelling in asthma: From benchside to clinical practice.Can Respir J. 2010 Jul-Aug;17(4):e85-93.
[2]Al-Muhsen S, Johnson JR, Hamid Q. Remodeling in asthma. J Allergy Clin Immunol. 2011 Sep;128(3):451-62; quiz 463-4.
[3]Cohen L, E X, Tarsi J, et al.Epithelial cell proliferation contributes to airway remodeling in severe asthma. Am J Respir Crit Care Med. 2007 Jul 15;176(2):138-45.
[4]Holgate ST.Epithelium dysfunction in asthma. J Allergy Clin Immunol. 2007 Dec;120(6):1233-44
[5]Hamilton LM, Puddicombe SM, Dearman RJ, et al.Altered protein tyrosine phosphorylation in asthmatic bronchial epithelium. Eur Respir J. 2005 Jun;25(6):978-85.
[6]Holgate ST. Innate and adaptive immune responses in asthma. Nature medicine,2012; 18 (5 ):673-83
[7]Puddicombe SM, Polosa R, Richter A, et al.Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J. 2000 Jul;14(10):1362-74.
[8]Puddicombe SM, Torres-Lozano C, Richter A, et al.Increased expression of p21(waf) cyclin-dependent kinase inhibitor in asthmatic bronchial epithelium. Am J Respir Cell Mol Biol. 2003 Jan;28(1):61-8.
[9] Davies DE. The Role of the Epithelium in Airway Remodeling in Asthma. Proc Am Thorac Soc. 2009 Dec;6(8):678-82.
[10] Manuyakorn W, Howarth PH, Holgate ST. Airway remodelling in asthma and novel therapy. Asian Pac J Allergy Immunol. 2013 Mar;31(1):3-10.
[11]Al-Alawi M, Hassan T, Chotirmall SH.Transforming growth factor β and severe asthma: a perfect storm. Respir Med. 2014 Oct;108(10):1409-23.
[12] Hackett TL, Warner SM, Stefanowicz D, et al. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1.Am J Respir Crit Care Med. 2009 Jul 15;180(2):122-33.
[13]Yasukawa A, Hosoki K, Toda M, et al.Eosinophils promote epithelial to mesenchymal transition of bronchial epithelial cells. PLoS One. 2013 May 21;8(5):e64281.
[14] Al-Muhsen S, Johnson JR, Hamid Q.Remodeling in asthma. J Allergy Clin Immunol. 2011 Sep;128(3):451-62
[15] Kudo M. Pathology of asthma. Front Microbiol. 2013 Sep 10;4:263.
[16] Chiba Y, Nakazawa S, Todoroki M,et al.Interleukin-13 augments bronchial smooth muscle contractility with an up-regulation of RhoA protein. Am J Respir Cell Mol Biol. 2009 Feb;40(2):159-67.
[17]Kudo M, Melton AC, Chen C, et al.IL-17A produced by αβ T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat Med. 2012 Mar 4;18(4):547-54.
[18] Mauad T, Xavier AC, Saldiva PH, et al. Elastosis and fragmentation of fibers of the elastic system in fatal asthma. Am J Respir Crit Care Med. 1999 Sep;160(3):968-75
[19] Bousquet J, Jeffery PK, Busse WW,et al. Asthma.From bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med. 2000 May;161(5):1720-45.
[20] Manuyakorn W, Howarth PH, Holgate ST.Airway remodelling in asthma and novel therapy. Asian Pac J Allergy Immunol. 2013 Mar;31(1):3-10.
[21] Schuliga M1, Javeed A, Harris T, et al.Transforming growth factor-β-induced differentiation of airway smooth muscle cells is inhibited by fibroblast growth factor-2. Am J Respir Cell Mol Biol. 2013 Mar;48(3):346-53.
[22] Pain M1, Bermudez O, Lacoste P, et al.Tissue remodelling in chronic bronchial diseases: from the epithelial to mesenchymal phenotype. Eur Respir Rev. 2014 Mar 1;23(131):118-30.
[23] Kumawat K, Koopmans T, Gosens R.β-catenin as a regulator and therapeutic target for asthmatic airway remodeling. Expert Opin Ther Targets. 2014 Sep;18(9):1023-34.
[24] Leclere M , Lavoie-Lamoureux A , Joubert P , et al . Corticosteroids and antigen avoidance decrease airway smooth muscle mass in an equine asthma model . Am J Respir Cell Mol Biol .2012 ; 47 ( 5 ): 589 – 596
[25] Hirota N, et al. Mechanisms of Airway Remodeling. Chest 2013; 144 ( 3 ): 1026 – 1032
[26] Kramer EL, Hardie WD, Mushaben EM, et al.Rapamycin decreases airway remodeling and hyperreactivity in a transgenic model of noninflammatory lung disease. J Appl Physiol (1985). 2011 Dec;111(6):1760-7.
[27] Sheshadri A, Castro M, Chen A.Bronchial thermoplasty: a novel therapy for severe asthma. Clin Chest Med. 2013 Sep;34(3):437-44.
[28] Chung KF, Wenzel SE, Brozek JL, et al.International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J 2014; 43: 343-373.
上一篇:
气道炎症标志物研究进展及其临床应用
下一篇:
职业性哮喘的诊断









